Genetics of Hypertrophic Cardiomyopathy: Key Points
19 July 2024
Genetics of Hypertrophic Cardiomyopathy: Key Points
The following are key points to remember from an article about genetics of hypertrophic cardiomyopathy (HCM), which reviews established and emerging implications for clinical practice:
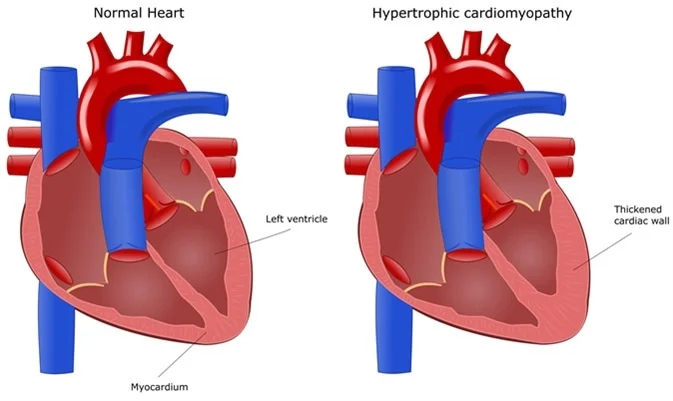
Most common genes identified in nonsyndromic, sarcomeric HCM include MYH7 (coding for beta-myosin heavy chain), MYBPC3 (myosin-binding protein C), TNNT2 (troponin T), and TNNI3(troponin I). These genes account for 90% of genotype-positive HCM cases.
Finding of a pathogenic or a likely pathogenic (P/LP) mutation in genes known to cause HCM improves diagnostic certainty. Accordingly, all guidelines recommend genetic testing for HCM.
Cascade genetic testing in relatives of patients with HCM who have a P/LP mutation identified can help define their risk of developing HCM and guide screening for the disease. Screening frequency in relatives depends on their age including annual testing in adolescence and early adulthood and every 3-5 years later in adulthood. Identifying a P/LP variant also allows for preimplantation genetic testing with in vitro fertilization.
Disease penetrance is higher among genotype-positive relatives of an individual with HCM, with male sex and presence of electrocardiogram abnormalities. TNNI3 has the lowest penetrance when compared to MYBPC3. Sudden cardiac death has not been noted in genotype-positive individuals without HCM phenotype.
In the absence of left ventricular hypertrophy (LVH), variant carriers can be noted to have diastolic dysfunction, fibrosis, myocardial crypts, enlongated mitral leaflets, myocardial perfusion defects, and electrophysiological abnormalities. If these findings are present, closer clinical screening for LVH development is recommended at a frequency of every 6-12 months.
Sarcomere-positive individuals with HCM present at an earlier age compared with sarcomere-negative individuals, have more severe hypertrophy, less frequent LV outflow tract obstruction, greater scar burden, and increased risk for arrhythmia and heart failure. However, genotype-phenotype associations have been clinically challenging to incorporate into risk prediction algorithms, as predictors include clinical factors such as age and maximal wall thickness that are at least partly correlated with genotype.
HCM phenocopies include Fabry disease, amyloidosis (TTR), PRKAG2 syndrome, Danon disease, and RASopathies such as Noonan syndrome. These diseases can be differentiated from age of onset, history, physical exam, presence of extracardiac symptoms, and variable mode of inheritance for some. Genetic testing differentiates across all these phenocopies, which has implications for clinical management (e.g., tafamidis for TTR amyloidosis).
Approximately 60% of HCM patients do not have an identifiable sarcomeric variant. Data suggest that development of HCM in these individuals may be influenced by environmental factors and polygenetic effects. Relatives of these individuals with “nonfamilial HCM” may not need to be screened as frequently as genotype-positive individuals and they may benefit from management of cardiac risk factors such as hypertension.
Disease-specific therapies are now available for HCM as cardiac myosin inhibitors including mavacamten and aficamten in this class. In trials of patients with symptomatic obstructive HCM, mavacamten and aficamten improved exercise capacity and symptoms compared with placebo. Another trial showed reduction proportion of patients that needed septal reduction therapy with mavacamten.
Gene therapy trials are in the early phases of testing for HCM and use adeno-associated vectors for DNA or lipid nanoparticles for RNA. Potential complications of gene editing therapy include off-target effects causing somatic cell mutagenesis and increasing the risk for cancer, virus vector immunogenicity, suboptimal delivery to the cardiomyocytes, and neutralization due to antibodies from previous adenovirus infections.